This “side-project” started off with a working title “Resolving the TNF Paradox”. We were puzzled why Tumor Necrosis Factor (TNF), a marker of high anti-parasite immune activity, is associated with high (rather than low) parasite densities in malaria. Superficially it makes sense, because parasite load is the main stimulus for the production of host defense cytokines. Indeed, many have postulated that excessive production of these cytokines contributes to severe malaria pathology. But the same cytokines also help with parasite clearance. So if a person produces a strong pro-inflammatory response to infection, how do they end up with high parasite load in the first place? Why don’t they control parasite load earlier, before high parasite load and severe disease develop? The longer we thought about the problem the more convinced we became that the “TNF paradox” likely arises from looking at “static” data that does not truly capture all aspects of the complicated dynamic interaction between the parasite and the host immune system.

A snap-shot of a tug of war (Photograph 519167, Record Group 75, National Archives at College Park, College Park, MD)
It’s the same problem we face in almost every observational study of severe infection in humans, where we only know what is happening at the time someone is ill enough to seek medical attention, and not what was happening before this. Without accounting for the dynamics of the interaction between host response and pathogen load over time, it’s like looking at a static picture of a tug of war. On one side is the rate of pathogen growth, and set against it is the host response, but the static picture provides no information about how hard each side is pulling, which way the rope is moving, or how close victory or defeat may be.
If only we could find a way of looking at the full dynamic course of malaria infection, parasite growth, and anti-parasite immune responses in patients, rather than being limited to data from one single snapshot in time. Obviously this information can’t be obtained directly from patients today because we have to give antimalarial treatment as soon as we make the diagnosis of this life-threatening infection. But we reckoned that there might be a way of reconstructing the time course of infection in patients, by making use of some unique historical data. This data comes from the pre-antibiotic era, from the 1920s onwards, when patients were deliberately infected to malaria to cure neurosyphilis. And yes, this so-called malariatherapy did actually work!
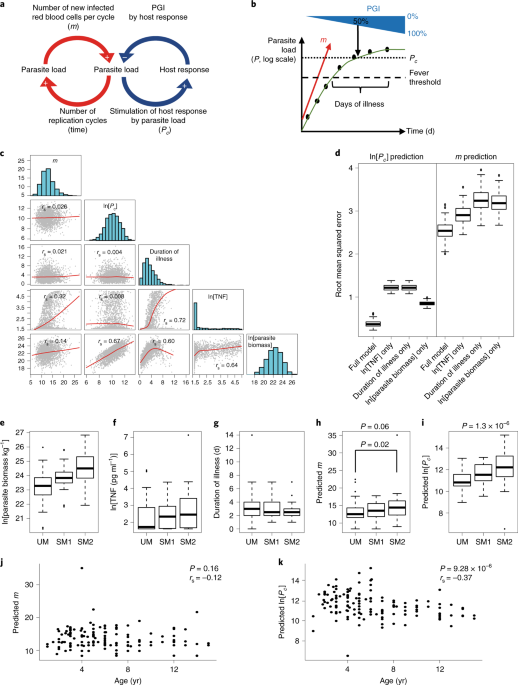
Sadly no one had the foresight to freeze down aliquots of plasma or RNA from these malariatherapy patients, but they did take meticulous records of parasitemia over time and noted the parasitemia necessary to cause fever in each individual. These data had already been summarized in the form of a mathematical model, a computer simulation focusing on the first wave of parasitemia, by Klaus Dietz and colleagues. Klaus kindly provided the individual-level parameter estimates necessary for us to re-implement the model, such that we would be able to simulate malaria patients. We had to make some adaptations to incorporate all the parameters of interest, but ultimately we were able to use it to simulate patients that resembled Gambian children we had recruited in a severe malaria study.
After a bit of playing around with the “clinical malaria simulator” and gaining an intuitive understanding of it, the idea came up that we might be able to estimate missing information on the unobserved course of infection in every individual Gambian patient by using the simulation outputs as reference map, together with some points of reference that were available in the data - a bit like the triangulation process used by orienteers (or mobile phones) to locate geographical position based on landmarks (or phone masts).
To do this, we trained a statistical model to learn from the simulations and predict the information that could not be directly observed in the Gambian patients (but was readily available in the simulation outputs). By doing this, we effectively oriented our map to the data from Gambian children collected at the time of presentation with malaria, and were able to estimate where each child was in the course of their infection. Not only could we do this, but we could estimate the opposing forces – how fast parasites were multiplying in each child and how strongly the each child’s immune response was opposing parasite growth. This provided a solution to our “TNF paradox” – the higher TNF levels being driven by faster replicating parasites and later presentation. However, we were stumped by the problem of how to prove the validity of our model. After all, the reason we had started this project was to try to measure something which couldn’t be directly measured, so how could it be validated?
It was pretty clear when we presented this at meetings that people were skeptical, so we decided to see if we could predict novel biology using our model. We combined the individual estimates of parasite growth inhibition with RNA-seq data to screen for the most strongly correlated gene expression. The resulting genes looked very interesting and suddenly the major focus of our work became mechanistic validation. To our surprise and delight we found that the two genes we selected for validation (Cathepsin G and Matrix Metallopeptidase 9), did indeed inhibit parasite growth. Even more exciting, we were able to characterize the mechanism of action of cathepsin G, which prevents parasite entry into host red cells by chopping off the red cell surface receptors needed for parasite invasion. Of course this still doesn’t prove beyond doubt that our triangulated estimates of parasite growth and host-response dynamics are accurate, but it shows that they are accurate enough to improve understanding of the process of host-parasite interaction and to predict novel mechanisms of protection.
So having started this project trying to resolve a paradox which seemed more important to us than to anyone else, five years later we have ended up developing an approach for understanding the dynamics of host-pathogen interaction in human malaria, which could have important applications. We can now extend this work to correlate any other measurements with parasite growth rate and parasite growth inhibition, in order to streamline the identification of their mechanistic determinants. This might include identification of genetic determinants, protective antibodies to inform vaccine design, or targets for host-directed therapy.



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in